AUCTORES
Globalize your Research

Case Report | DOI: https://doi.org/10.31579/2578-8949/016
*Corresponding Author: Julieta Ruiz Beguerie, MD Austral University, Austral University Hospital, Dermatology Department. Av Peron 1500. Pilar, Buenos Aires Province. Argentina
Citation: Ruiz Beguerie Julieta, Marchese Maria Laura, Anaya Javier, Busso Corina. (2018) A Rare Presentation of Cutaneous Plasmocytosis in a Caucasian Woman . J Dermatology. DOI:10.31579/2578-8949/016
Copyright: © 2018 Marchese, Maria Laura1, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 11 December 2017 | Accepted: 05 January 2018 | Published: 10 January 2018
Keywords: Cutaneous plasmocitosis; Plasma cells; Polyclonal hypergammaglobulinemia
Cutaneous plasmocytosis is a rare disorder mainly occuring in Japanese people, characterized by a polyclonal plasma cell infiltration in the skin. Sometimes this infiltration can also be found in other organs, which in that case it is called Systemic Plasmocytosis. We report the first case of a 44- year -old caucasian woman without relevant medical history , with manifestations of the disease only in the oral mucous without expressing lesions on the trunk , where it usually appears. Histopathological examination revealed a perivascular infiltrate composed predominantly of plasma cells expressing kappa and lamba light chains. Laboratory test demonstrated polyclonal hypergammaglobulinemia. The patient responded to oral prednisone and tea tree oil. This rare case highlights the spectrum of clinicopathologic presentations which, may be wider than previously recognized.
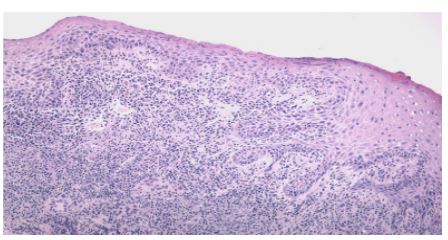
A woman of 44 years old, mother of two healthy siblings, presented to our clinic with a six-year history of a non-pruritic erythematous plaque on the upper lip which compromises the oral mucous underneath where an ulcer can be seen. (Fig 1 and 2)
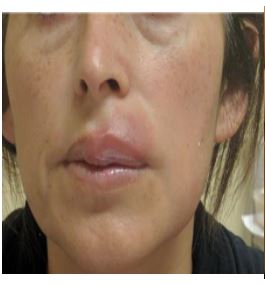
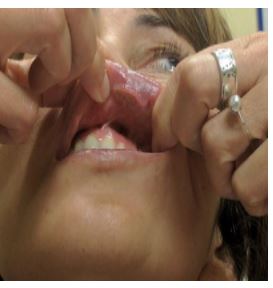
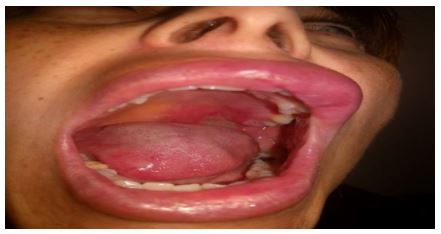
The patient had no prior medical history and did not take any medication. She specifically denied fever, weight loss, fatigue, dyspnea, abdominal pain, and changes in urination. She had not attempted any topical or oral treatment prior to the visit .The patient referred to be slightly painful when touching the lesion. She also had a plaque on the right lateral portion of the tongue and erosion on the left side of the hard palate. (Fig 3) There were no papules or plaques or tumors on the rest of the skin.
Skin biopsies were done of the plaque and ulcer in the oral mucous close to the upper lip (Fig 4 &5).

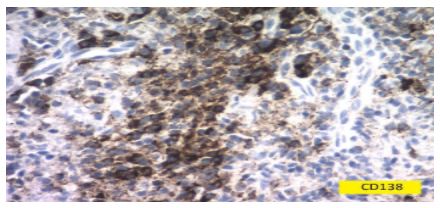
The stain CD138 in plasma cells of the skin biopsies were positive (Fig 6).We also asked for laboratory tests, including VDRL, and serum proteins.
The tongue and oral mucous swabs came back negative to possible infections.
After receiving the results from the lab and the pathology report suggesting a cutaneous plasmocytosis, we decided to go further beyond and explore a systemic plasmocytosis with CT scan from thorax, abdomen and pelvis. These last tests came back normal. Lymphadenopathy in the right side of the neck was detected by ultrasound. Her laboratory showed the expected polyclonal expression of immunoglobulin light chain by the plasma cells.As her physical cexamination, laboratory tests, and radiographic examination were negative for systemic involvement we diagnosed cutaneous plasmocytosis with only oral manifestation . To our knowledge this is the first case with only manifestations in that location in a caucasian woman.
So, we decided to start with oral steroids in a low dose (30mg per day of prednisone) and close follow up. The patient had begun adding tea tree oil to the oral lesions with moderate relief.
In 1976, Dr Yashiro first reported a case of this disease under the title “A kind of plasmocytosis”, and later termed cutaneous plasmocytosis was given by Kitamura and colleagues [1, 2]. The geographic distribution of disease cases with the main point in Japan led to different presumptions that it might be the result of environmental factors, genetics and infectious disease occurring in that area such as borreliosis, syphilis or HHV-8. No molecule mutation has been detected so far no evidence of infectious triggers [3, 4]. The fact that IL-6 mRNA is detected only in skin lesions of plasmocytosis and the observation that plasma cells express IL-6, it is hypothesized that there is a cellular defect in the plasma cells themselves which might lead to an autocrine-paracrine stimulation of the cells and as a consequence of this phenomenon, skin or systemic manifestations appear [5].
Cutaneous plasmocitosis usually presents clinically with multiple, symmetrical, red-brown infiltrated plaques and flat tumors, mainly located on the trunk, face and nape. Only 40% of the patients manifest pruritus which is usually moderate [6, 7].
Etiology and pathogenesis are not well known. It is speculated that a reactive dysfunction of plasma cells may be triggered by multiple factors, such as interleukin [6, 5].
Histopathologic characteristics show marked hyperplasia of mature polyclonal plasma cells without atypia located at dermal superficial and deep level mainly perivascular, and polyclonal hypergammaglobulinemia on serum protein electrophoresis desecrating secondary cause. Frequently, the infiltrate is also found perineurally and occasionally plasma cells are found in the nerve fascicles. In 33% of the cases there can be found small reactive germinal centers [6].
Apart from skin lesions, polyclonal hypergammaglobulinemia is the most frequent manifestation of cutaneous plamocytosis, seen in 88–93 % of the patients [6]. Most proliferations are IgG and IgA. IgM is distinctly rarer and less pronounced [8]. The importance of this observation is that the level of the hypergammaglobulinemia is viewed as a prognostic factor by Uhara and collaborators. Extra cutaneous involvement and systemic signs are more common in patients with IgG levels over 5,000 mg/dl [6]. A concentration of plasma cells of more than 7 % has just the same importance than an IgG value of over 5,000 mg/dl. Both values are viewed as a prognostic indication of a more severe disease course of plasmocytosis [6].
Adenopathies are detected in half of the patients with cutaneous plasmocytosis diagnosis according to the cases reported in the literature [9].
Until 2013, there were only 67 cases reported in the literature which only 10 were observed in Caucasian patients [10]. The rest were all Asian, Japanese, Chinese, Thais and Koreans. It is mainly reported in adults, between the third and the sixth decade although there are few reports in children [11]. The male/ female ratio is found to be 1:0.6 [6]. It is also reported in dogs, especially in breeds such as Golden and Labrador retriever, with pink alopecic or ulcerated tumors in the trunk with or without systemic compromise [12].
It is a rare entity arising primarily in patients of Japanese descent, which was not the case in our patient. The exact pathogenesis is poorly understood.
The third most frequent place of involvement apart from skin and lymph nodes is the bone marrow, which was detected positive in 40% of the patients studied by Shimizu and colleagues in 1997 [13].
When systemic plasmocytosis is suspected, it can be found hepatosplenomegaly, interstitial pneumonia and nephropathies together with fatigue, fever or weight loss [13].
The differential diagnoses to take into account in a cutaneous plasmocytosis are: syphylis, borreliosis, systemic plasmocytosis, cutaneous plasmocytoma, Casstleman’s disease, cutaneous leichmaniasis, mastocytosis, lichen planus, pytiriasis rosacea, parapsoriasis, lymphoma, leukemia cutis and lupus erythematous. Myeloproliferative neoplasias and inflammatory dermatoses must be excluded.
Systemic Plasmacytosis (SP), which has a histologic appearance similar to that of multicentric Castleman's disease (MCD), is also known as benign plasma cell proliferation with polyclonal hypergammaglobulinemia, cutaneous plasmacytosis, and/or generalized plasmacytic lymphadenopathy.
The general symptoms of this entity are unspecific and include fatigue, weight loss, fever and dyspnea(14).The prognosis of SP reportedly has been good in most cases although there are reports of development of a T- cell lymphoma in some patients [2].
Cutaneous Solitary Plasmacytoma with no evidence of systemic disease is a rare disorder comprising 5%-10% of all plasma cell neoplasms. Progression to multiple myeloma is the most common pattern of relapse [15].
Treatment of cutaneous plasmacytosis is difficult with little response or a slight benefit is seen. The treatments proposed for this rare entity are not well stablished but the the literature mentioned the following: PUVA, topical corticosteroids and calcineurin inhibitors (tacrolimus or pimecrolimus), oral antibiotics or oral steroids (30-40mg prednisolone, daily) and melphalan are mainly used [16, 17 & 18].
If pruritus is an important issue for the patient, it can be relieved by pimecrolimus with daily application [19]. Some reports in the literature mentioned a complete spontaneous remission in the following year of the diagnosis [4].
Conclusion
The differentiation between cutaneous and systemic plasmocytosis appears problematic in practice many times. Most patients diagnosed at the beginning as cutaneous plasmocytosis, can actually have an occult systemic manifestation, which is not yet expressed at the time of first diagnosis, so it is recommended to follow up closely these patients even after remission of their cutaneous lesions [9]. The clinical presentation of our patient deviated from the typical appearance suggesting that the spectrum of clinicopathologic presentations may be wider than previously recognized.
Journal of Clinical Cardiology and Cardiovascular Intervention The submission and review process was adequate. However I think that the publication total value should have been enlightened in early fases. Thank you for all.